

Neurofibromatosis Type 1
Overview
Neurofibromatosis Type 1 (NF1) is the most common type of NF and is also one of the most common inherited neurological conditions affecting about 1 in 2500 people worldwide NF1 causes unchecked growth of tissue along the nerves.
Approximately 50% of individuals diagnosed with NF1 inherit NF1 from a parent the other 50% will be the first person in their family to have the variant or change in the NF gene There is no way to predict the severity of NF1 Severely affected parents may have mildly affected children and mildly affected parents may have severely affected children.
50
%
NF1 is a very unpredictable disorder It varies widely in severity from one person to the next even between two people in the same family Some people with NF1 go through life with only a few café-au-lait spots or neurofibromas while others may have major cosmetic or medical problems due to NF1 Although NF 1 is a very unpredictable disorder and varies widely in severity from person to person we have witnessed an amazing spirit with in NF individuals who create It also should not limit the future for individuals with NF
The upcoming content will contain information to help you understand some of the manifestations, diagnostic criteria, treatments, genetics and other helpful resources
Manifestations
Below are the potential symptoms of NF1
Skin
- Cafe-au-lait spots
- Freckling in skin creases
- Severe itching
Tumor
- Neurofibromas
- Plexiforms
- Brain Tumors
Cognitive
- Seizures
- ODD
- ADHD
- Autism
- Brain Blood Vessel Defects
- Macrocephaly
- Psychosocial burdens
Visual
- Visual Impairment/Blindness
- Optic Glioma
- Lisch Nodules
- Migraine Headaches
Speech
- Speech Impairments
Cardiovascular
- High Blood Pressure
- Cardiovascular Disease
- Inflammation
Digestive
- Digestive Issues
- Chronic Constipation
- Diarrhea or Vomiting
Skeletal
- Scoliosis
- Pseudoarthrosis
- Bone Deformities
- Short stature
Cancer
- Cancer
- Breast and Other Cancers
Chronic Pain
- Chronic Pain
Important Note
While there are many manifestations that can occur within NF, no one person will have all have the manifestations listed above.
Diagnostic Criteria
A diagnosis of NF1 is made when an individual has two or more of the following findings:
Café au lait spots
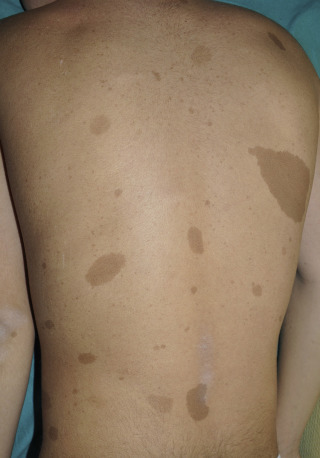
birth or develop during the first few years of life. Café-au-lait spots are not dangerous but indicate the possible presence of an NF1 gene change in the person. At least one of the two pigmentary findings (café-au-lait or freckling) should be bilateral.
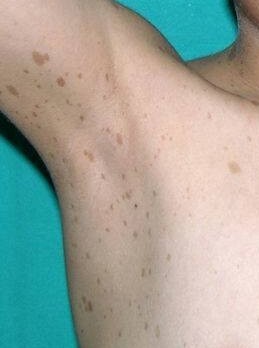
Freckling
Freckling in the armpits or the groin. This usually appears by 3 to 5 years of age. Freckles are similar in appearance to café-au-lait spots but are smaller in size. At least one of the two pigmentary findings (café-au-lait or freckling) should be bilateral.
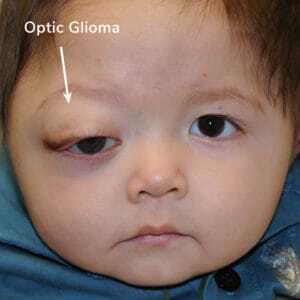
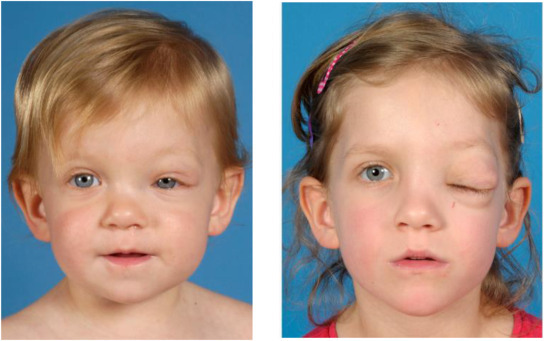
Optic glioma
A tumor of the optic pathway (optic pathway glioma). These tumors typically first appear
by age 6, rarely in late childhood and adolescence, and almost never in adults. Although they can affect vision, most do not become symptomatic.
Family history of NF1
A parent, sibling, or child with NF1.
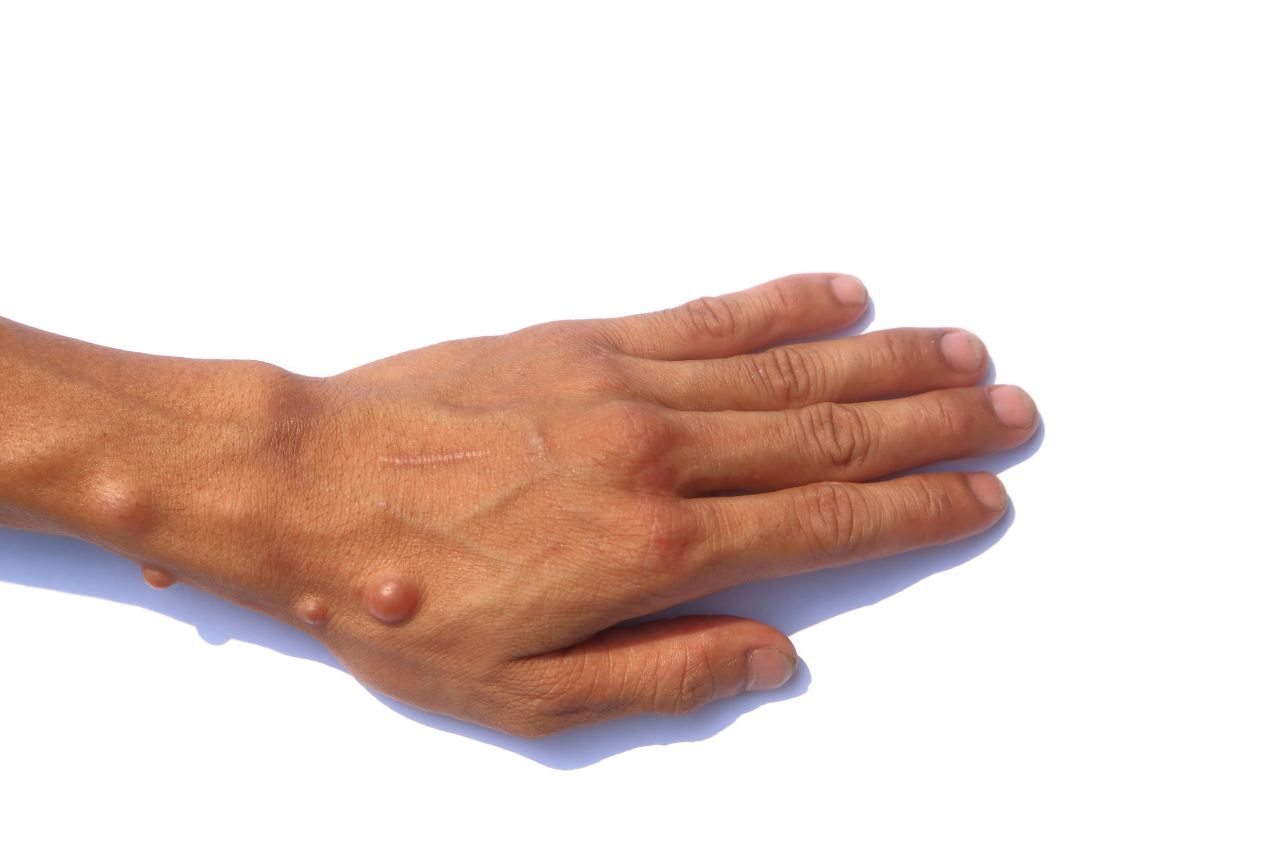
Neurofibroma or Plexiform

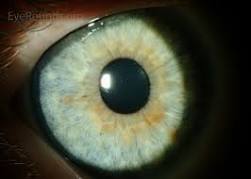
Lisch Nodule
Two or more growths on the iris of the eye (known as Lisch nodules or iris hamartomas). These nodules are harmless, not typically present until adolescence, do not affect vision, and do not require monitoring or treatment.
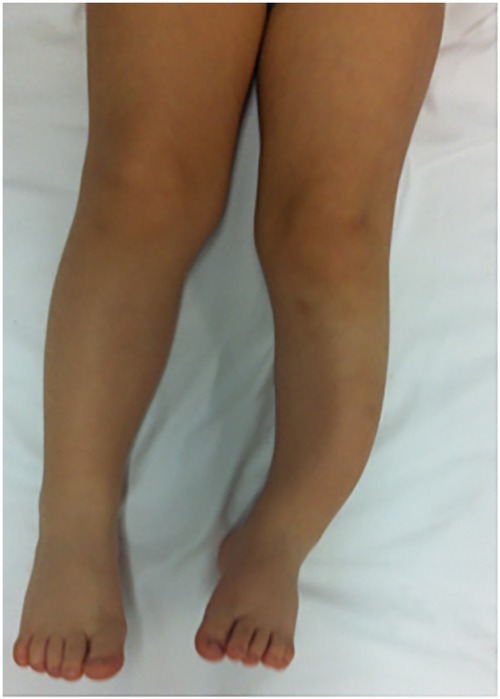
Bone abnormalities
Abnormal development of the eye socket (sphenoid) or the tibia (one of the long bones of the shin).
Genetic Testing
Genetic testing is not REQUIRED for diagnosis but may allow for an earlier diagnosis. Genetic testing ALONE is not sufficient to diagnose NF1 – diagnosis requires a 2nd diagnostic feature of NF1. Genetic testing cannot predict the severity or specific complications in any individual case of NF1.
Treatments
Neurofibromatosis is a complicated condition, and it should be treated at a major medical center by specialists with expertise in the condition. An individual with neurofibromatosis is best treated by an interdisciplinary team that may include the following:
- Neurologist
- Geneticist
- Dermatologist
- Cardiologist
- Ophthalmologist
- Hearing Specialist
- Physical, Occupational, and/or Speech Therapist
- Orthopedist
- Neurosurgeon
- Oncologist
- Psychologists
Treatments for neurofibromatosis are symptomatic, meaning that they treat the symptoms but do not cure the disease. Individuals with neurofibromatosis may be treated with medicine, pain management techniques, various therapies, and surgery, as symptoms arise.
The NF Network offers a tool to help find an experienced NF clinical team. Our office is available for consultation if the need arises.
If access to an NF Clinic is not available in your area our Clinical Care Options publication may help bridge the gap of care between your local physician and an experienced practitioner. We have published a comprehensive scientific publication created for the express purpose of improved clinical care. Designed for both the patient and the local physician, often with limited NF knowledge, it includes information on the diagnosis and development of NF, specific tumor types, and a list of Neurofibromatosis Clinical Trials Consortium clinics whose practitioners, collectively are up to date on available treatments and who have agreed to be contacted by physicians to learn more about NF or to refer a patient.
Download the Clinical Care Options GuideGenetics
Neurofibromatosis type 1 (NF1) is a hereditary disorder caused by a change in the NF1 gene, which is located on chromosome 17. More than 1,000 NF1 mutations that cause neurofibromatosis type 1 have been identified. (Image if possible)
The NF1 gene makes a protein called neurofibromin, which regulates cell division in the nervous system and functions as a kind of molecular brake to keep cells from growing out of control. Ongoing research continues to discover additional genes and molecular pathways that may play a role in NF-related tumor suppression or growth. Continuing research is starting to reveal how this novel family of growth regulators controls how and where tumors form and grow, which may lead to the development of new drugs and therapies for NF.
Current basic and clinical research is not only aimed at understanding how genetic defects cause the diverse conditions and medical problems encountered by people with NF, but also how to predict which clinical features will arise in any given person (personalized or precision medicine).
Inheritance VS Spontaneous
All people have two copies of every gene – one copy inherited from each parent. The NF1 gene mutation is dominant, which means that only one of the two copies of the gene needs to have the mutation to produce the disorder. A parent with NF1 has a 50% chance of passing the abnormal gene copy to a child. A child who inherits the altered gene will also have the disorder.
While half of the cases of NF1 are inherited from a parent, 50% of children diagnosed with NF1 appear to be the first members of their family to have the disorder. In such cases, the genetic alteration, or mutation, occurred in the sperm or egg cell that formed the child. This is called a spontaneous, or new, mutation. A person with a spontaneous mutation of the NF1 gene has a 50% chance of passing the abnormal gene copy to a child.
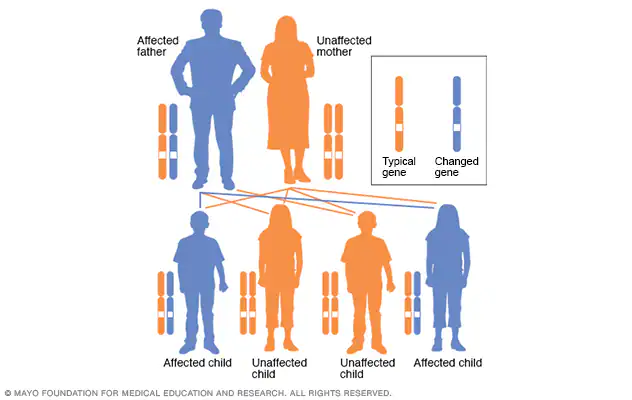
In cases where a child’s NF1 is determined to be the result of a spontaneous mutation, parents often wonder if they did something to cause the mutation, such as exposing their child to radiation, medications, alcohol, or any other substances in the environment. It’s important to understand that the specific cause of the NF1 gene mutation is currently unknown, and no environmental exposure has been implicated. It’s also important to know that genetic mutations are not uncommon. Cells in the body are continuously dividing, and each time they do, a massive volume of genetic information must be copied correctly. Random errors inevitably occur in the copying process and may be the cause of mutations leading to NF1.
Segmental or Mosaic
Individuals with segmental NF have only a certain area of the body that shows signs of NF. Any part of the body can be affected.
Segmental NF1 – Individuals with segmental NF1 most commonly have the skin findings associated with NF1, such as café au lait spots, skin fold freckles, or neurofibromas, that are confined to a certain area (for example, only an arm or only a restricted area of the abdomen). Some individuals may have an isolated tumor involving the nerves without other signs of NF1. It is not known whether such isolated tumors represent true cases of segmental NF, or whether they may have other causes.
Segmental NF2 – Individuals with segmental NF2 have some of the tumors associated with NF2 confined to a restricted region, such as only one side of the body.
Segmental Schwannomatosis – Individuals with segmental schwannomatosis have tumors associated with schwannomatosis confined to a restricted region, such as only one limb or a part of the spine.
Microdeletion

Approximately 5% of individuals with a diagnosis of NF1 have a deletion that includes the entire NF1 gene. When the entire NF1 gene is missing, it is referred to as an NF1 (whole) gene deletion. Some people also call this an NF1 microdeletion. The deletion may include other nearby genes. Some of the surrounding genes are important for cognitive functioning and body development. Individuals with an NF1 gene deletion are often, but not always, characterized by a more severe presentation than is observed in other individuals with NF1.
Individuals with the NF1 gene deletion often have earlier and more numerous neurofibromas, as well as an increased risk of transformation of plexiform neurofibromas into a malignant/cancerous form of a tumor. Cardiac malformations and scoliosis are also more common in individuals with a whole NF1 gene deletion. In addition, most individuals with an NF1 gene deletion have more significant learning and developmental issues, and intellectual disability may be present.